Hydrogen Deuterium Exchange (HDX) Mass Spectrometry Service
- Service Details
- Case Study
Hydrogen-deuterium exchange mass spectrometry (HDX-MS) is a mass spectrometry technique that uses the proton instability mass present on the amide of the protein backbone to study the protein structure. The principle is that when a protein is immersed in a solution of deuterium water, the amide hydrogens in the main chain of the protein molecule will exchange with the deuterium in solution at different rates, depending to some extent on the conformation. The hydrogen on the surface of the protein in close contact with the deuterium water has a faster rate of hydrogen-deuterium exchange than that formed by the hydrogen located inside the protein. The rate of hydrogen-deuterium exchange for different sequence fragments of the protein is determined by mass spectrometry to derive information about the protein spatial structure and to infer the degree of activity at different sites.
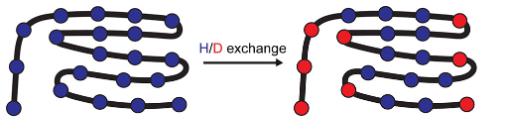
The benefits of HDX-MS include: independence from the size and complexity of the analytical system; low sample requirements; high sensitivity; and the ability to detect coexisting protein conformations.
Creative Proteomics offers a wide range of solutions for hydrogen-deuterium exchange mass spectrometry (HDX-MS) projects. We are equipped with the nanoACQUITY UPLC HD-Exchange System analytical system with sub-dimensional nano-chromatographic particle packing for ultra-high separation. The on-line automated pre-treatment is used inline with the high-resolution mass spectrometer to improve experimental efficiency and fully guarantee the effectiveness of pre-treatment and reliability of identification results.
Creative Protomics's HDX-MS service can provide:
- Epitope mapping
- Identify drug binding sites and drug-induced conformational changes
- Analyze protein-protein interactions
- Provide information on protein aggregation, conformational changes and regions of intermediate structures formed during aggregation
- Study the effects of mutations on protein structure and dynamics
- Study the effect of agents on the structure or stability of biological drugs
- Analyze information on conformational differences between different states of proteins or protein complexes and elucidate structured versus unstructured regions of proteins
- Study the effects of phosphorylation and other post-translational modifications on protein structure and dynamics
Workflow of HDX-MS
 General workflow for HDX-MS experiments (Masson et al., 2013)
General workflow for HDX-MS experiments (Masson et al., 2013)
Sample Delivery Requirements
- Sample type: purified protein or molecule/antibody of known sequence and known binding conditions
- Total sample volume: ≥ 0.10mg; > 0.05mg (antibody/antigen Epitope mapping)
- Sample concentration: ≥ 0.5mg/mL
- Sample purity: ≥ 90%
If you would like to learn more about HDX-MS services or have other needs, please contact us. We look forward to cooperating with you.
Reference
- Masson, G. R., Burke, J. E., Ahn, N. G., Anand, G. S., Borchers, C., Brier, S., ... & Rand, K. D. (2019). Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nature methods, 16(7), 595-602.
Case: Structural Dynamics of Apolipoprotein-D Revealed Through Hydrogen-Deuterium Exchange Mass Spectrometry
Background
Apolipoprotein-D (apoD) is a glycosylated protein implicated in various biological functions, including lipid transport and neuroprotection. Understanding its structural dynamics, especially in the ligand-binding pocket, is crucial for unraveling its functional mechanisms. Hydrogen-deuterium exchange mass spectrometry (HDX-MS) provides a powerful tool to investigate the conformational changes in apoD, particularly in response to ligand binding.
Samples
The study utilized native, tetrameric, glycosylated apoD purified from human breast cyst fluid. Ligand-free and progesterone-bound apoD samples were subjected to HDX-MS to probe structural alterations.
Technical Methods
1. ApoD Purification:
- ApoD was purified from breast cyst fluid using anion exchange chromatography (ANX Sepharose FF) and size exclusion chromatography (Superdex 200 16/600).
- Fractions containing apoD were identified by Coomassie SDS-PAGE, pooled, and concentrated.
- ApoD concentration was measured using a Pierce BCA assay.
2. Amide Hydrogen-Deuterium Exchange:
- Deuterated buffer was prepared by vacuum-drying phosphate buffer and reconstituting with deuterium oxide.
- Deuterium exchange was performed on ligand-free and progesterone-bound apoD in triplicates.
- Reactions were quenched with a solution containing trifluoroacetic acid, GuHCl, and TCEP before injection into the LC–MS system.
3. Liquid Chromatography-Mass Spectrometry (LC–MS):
- A nanoACQUITY UPLC system and SYNAPT G2-Si mass spectrometer were used.
- Peptides were separated on a C18 reversed-phase column with a water-acetonitrile gradient.
- Peptides were detected using a SYNAPT G2-Si mass spectrometer in MSE mode.
- Data analysis involved searching against a human mature apoD database and calculating relative fractional uptake.
4. Data Analysis and Visualization:
- Graphs were created using GraphPad Prism 7, depicting standard deviations.
- A significant difference in absolute deuterium uptake of > 0.5 Da at least for one time point was considered.
- Protein structures were visualized using PyMOL, aligning structures from ligand-free and progesterone-bound models.
Results
Structural Insights into Ligand-Free ApoD:
- Ligand-free apoD displayed a stable lipocalin fold, with low deuterium exchange indicating a highly structured protein.
- Peptides in the β-barrel and adjacent α-helix regions showed limited dynamics, emphasizing the rigid nature of apoD.
Progesterone Binding Induces Structural Stabilization:
- HDX-MS revealed significant differences in deuterium exchange between ligand-free and progesterone-bound apoD.
- Peptides in the ligand-binding pocket exhibited reduced deuterium exchange, indicating stabilization upon progesterone binding.
- Allosteric effects were observed in distal regions, including the N- and C-termini.
Implications for ApoD Function:
- Stabilization of crucial regions, including the ligand binding pocket, upon progesterone binding may impact apoD's role in ligand binding, membrane interaction, and release dynamics.
- Allosteric changes in the N-terminal peptide suggest potential implications for apoD's interaction with its receptor, basigin.
 Relative fractional deuterium exchange of the apo-form of apoD reveals differences in exchange and dynamics.
Relative fractional deuterium exchange of the apo-form of apoD reveals differences in exchange and dynamics.
 Overview of exchange and dynamics over the apoD structure. (A) Relative fraction deuterium exchange after 120 min of non-overlapping peptides in putty representation and colour-coded. (B) Areas with low (area 2, magenta) and high dynamics (areas 1 and 3, green).
Overview of exchange and dynamics over the apoD structure. (A) Relative fraction deuterium exchange after 120 min of non-overlapping peptides in putty representation and colour-coded. (B) Areas with low (area 2, magenta) and high dynamics (areas 1 and 3, green).
Reference
- Kielkopf, Claudia S., et al. "HDX‐MS reveals orthosteric and allosteric changes in apolipoprotein‐D structural dynamics upon binding of progesterone." Protein Science 28.2 (2019): 365-374.
